
Progress in Mind
Amyloid precursor protein/amyloid-beta and tau appear to act together to cause deficits in synaptic function and neural hypersynchrony, which leads to cognitive dysfunction in Alzheimer’s Disease. Understanding the underlying mechanisms of this cascade could reveal novel therapeutic strategies.
Neural network hypersynchrony and synaptic depression are likely to be promoted by neuronal loss and contribute to neurodegeneration and cognitive decline in Alzheimer’s Disease (AD).1 In his plenary lecture, Lennart Mucke, MD (Gladstone Institute of Neurological Disease, USA) examined the impact of the underlying pathophysiology of AD on neural network function.
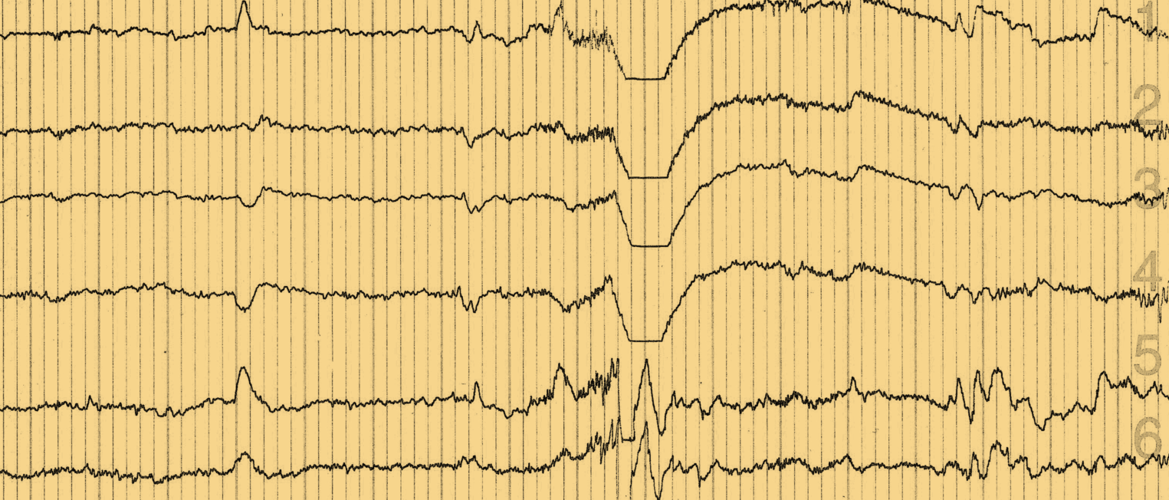
Non-convulsive epileptic activity is common in AD and is associated with the onset of cognitive decline
In amyloid precursor protein (APP) transgenic mice, decreased synaptic transmission is seen alongside network excitability, which is apparent as both convulsive and non-convulsive epileptic activity. In the absence of endogenous tau, EEGs appear normal, suggesting that amyloid-beta (Aβ) requires tau to induce hypersynchrony.
APP C-terminal fragments are elevated in patients with autosomal dominant AD; and many patients with familial AD mutations experience clinically observed seizures. 1, 2 In patients with mild cognitive impairment (MCI) or sporadic AD and epilepsy, a high proportion of seizures (>50%) are non-convulsive and seizure onset often clusters around the beginning of cognitive decline.3 Subclinical epileptic activity is detected in more than 40% of patients with early-onset, sporadic AD and appears to be associated with more rapid cognitive decline.3, 4 Other studies have also shown that measures of network dysfunction are useful biomarkers for AD.5-7
The degree of epileptic activity in patients with AD seems to be greatest at night, when EEG detection is also most sensitive, with 1‑hour of sleep recording equivalent to 8‑hours of daytime recording.8, 9 Magneto-encephalography with EEG also appears to be more sensitive to subclinical epileptic activity in patients with sporadic AD than 24-hour video EEG.4
SV2A-binding antiepileptic drugs can block the network dysfunction cascade
Antiepileptic drugs that bind the synaptic vesicle protein 2A (SV2A) have been shown to reverse hippocampal hyperactivation in the dentate gyrus/CA3 and improve performance on a pattern separation task in patients with amnestic MCI.10
APP/ Aβ, ApoE4 and tau play a role in synaptic and network dysfunction
In people with epilepsy, fast gamma oscillations are associated with the suppression of epileptic activity. Mouse and human studies of AD suggest that APP/Aβ impair fast gamma oscillations by depleting interneuron NaV1.1 sodium channels, leading to hypersynchrony and decreased cognitive function.11, 12 In contrast, restoring NaV1.1 activity in mice has shown benefits on brain dysrhythmias and cognitive deficits, while increasing gamma oscillations led to prevention of plaque load. 11, 12
ApoE4 and Tau may also play a role in hypersynchrony and subsequent cognitive impairment:
However a number of questions remain with regard to the best therapeutic approach. We do not know which is the most pathogenic form of tau, nor whether spreading of tau impairs critical neuronal functions or survival. Reductions in the power of delta/theta oscillations are seen in tau-ablated mice – and increases in delta/theta power predict progression to AD in people with subjective impairment.16, 17